Your Location:Home >Products >Organic phosphines >CyclohexyI phosphines >2622-14-2
Product Details
Description
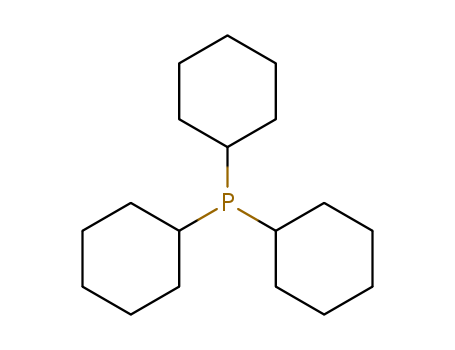
Tricyclohexyl phosphine is the tertiary phosphine with the formula P( C 6H11) 3. Commonly used as a ligand in organometallic chemistry, it is often abbreviated to PCy 3, where Cy stands for cyclohexyl. It is characterized by both high basicity (pK a = 9.7) and a large ligand cone angle (170°). Tricyclohexyl-phosphine is more basic (and so a better c-donor) than tribu tyiphosphine, which in turn is more basic than triphenyl phosphIne.
Chemical Properties
clear colorless to pale yellow solution
Uses
Tricyclohexyl Phosphine is a ligand catalyst used primarily in organometallic chemistry. Ligand used in the Pd-catalyzed coupling of malononitrile with aryl halides. suzuki reaction: Bulky phosphine ligand used with a Pd(0)-triolefinic macrocycle catalyst for Suzuki coupling of aryl bromides and chlorides. This ligand is applied with Ni as a key intermediate in the formation of cylcopentane compounds. As a reagent, Tricyclohexylphosphine is used for a range of metal-catalyzed organic transformations. Crabtree's catalyst is an organoiridium compound containing tricyclohexyl phosphine used for hydrogenation of mono-, di-, tri-, and tetra- substituted substrates and hydrogen transfer reactions. Grubbs' catalysts consists of transition metal ruthenium and tricylohexyl phosphine utilized in olefin metathesis in synthetic organic chemistry.
Uses
Tricyclohexylphosphine is used as a ligand in organometallic chemistry. As a reagent, it is used for a range of metal-catalyzed organic transformations. Crabtree's catalyst is an organoiridium compound containing tricyclohexyl phosphine used for hydrogenation of mono-, di-, tri-, and tetra- substituted substrates and hydrogen transfer reactions. Grubbs?catalysts consists of transition metal ruthenium and tricylohexyl phosphine utilized in olefin metathesis in synthetic organic chemistry.
Purification Methods
It recrystallises from EtOH [Boert et al. J Am Chem Soc 109 7781 1987]. [Beilstein 16 IV 947.]
InChI:InChI=1/C18H33P/c1-4-10-16(11-5-1)19(17-12-6-2-7-13-17)18-14-8-3-9-15-18/h16-18H,1-15H2
Sterically accessible Lewis donors are shown to accelerate decomposition during catalysis, for a broad range of Grubbs-class metathesis catalysts. These include benzylidene derivatives RuCl2(NHC)(PCy3)(=CHPh) (Ru-2: NHC = H2IMes, a; IMes, b; H2IPr, c; IPr, d; H2ITol, e) and indenylidene complexes RuCl2(NHC)(PCy3)(=C15H10) (NHC = H2IMes, Ru-2f; IMes, Ru-2g). All of these precatalysts form methylidene complex RuCl2(NHC)(=CH2) Ru-3 as the active species in metathesis of terminal olefins, and generate RuCl2(NHC)(PCy3)(=CH2) Ru-4 as the catalyst resting state. On treatment with a 10-fold excess of pyridine, Ru-4a and Ru-4b decomposed within minutes in solution at RT, eliminating [MePCy3]Cl A by net loss of three ligands (PCy3, methylidene, and one chloride), and a mesityl proton. In comparison, loss of A from Ru-4a in the absence of a donor requires up to 3 days at 55 °C. The σ-alkyl intermediate RuCl2(13CH2PCy3)(NHC) (py)2 resulting from nucleophilic attack of free PCy3 on the methylidene ligand was undetectable for the H2IMes system, but was spectroscopically observable for the IMes system. The relevance of this pathway to decomposition of catalysts Ru-2a-g was demonstrated by assessing the impact of pyridine on the in situ-generated methylidene species. Slow initiation (as observed for the indenylidene catalysts) did not protect against methylidene abstraction. Importantly, studies with Ru-4a and Ru-4b indicated that weaker donors (THF, MeCN, DMSO, MeOH, and even H2O) likewise promote this pathway, at rates that increase with donor concentration, and severely degrade catalyst productivity in RCM, even for a readily cyclized substrate. In all cases, A was the sole or major 31P-containing decomposition product. For DMSO, a first-order dependence of decomposition rates on DMSO concentration was established. This behavior sends a warning about the use of phosphine-stabilized metathesis catalysts in donor solvents, or with substrates bearing readily accessible donor sites. Addition of pyridine to RuCl2(H2IMes)(PCy3)(=CHMe) did not result in ethylidene abstraction, indicating that this decomposition pathway can be inhibited by use of substrates in which the olefin bears a β-methyl group.
An unsymmetrical PNP-pincer-type phosphaalkene ligand, 2-(phospholanylmethyl)-6-(2-phosphaethenyl)pyridine (PPEP), has been prepared from 2,6-bis(2-phosphaethenyl)pyridine (BPEP) by intramolecular C-H addition/cyclization of the 2-phosphaethenyl group with a 2,4,6-tri-tert-butylphenyl substituent (CH=PMes). The reaction proceeds in hexane in the presence of a catalytic amount of [Pt(PCy3)2] (20 mol %) at 80 C in a sealed tube, giving PPEP in 32% isolated yield, along with byproduction of 2,6-bis(phospholanylmethyl)pyridine (BPMP) and a Pt(II) phosphanido complex (5). The PPEP ligand reacts with [Rh(μ-Cl)(C2H4)2]2 and [RuCl2(PPh3)3] to afford [RhCl(PPEP)] (6) and [RuCl2(PPh3)(PPEP)] (8), respectively. Complex 6 easily undergoes C-H addition/cyclization at the other CH=PMes group to afford the 2,6-bis(phospholanylmethyl)pyridine complex [RhCl(BPMP)] (7), whereas 8 is stable against C-H addition/cyclization. Treatment of 8 with tBuOK forms [RuCl(PPh3)(PPEP)] (9), coordinated with an unsymmetrical PNP-pincer-type phosphaalkene ligand containing a dearomatized pyridine unit (PPEP). The X-ray structures of 5 and 9 are reported. The reaction processes from BPEP to PPEP and to 5 are discussed based on NMR observations.
The kinetics of quinuclidine displacement of BH3 from a wide range of Lewis base borane adducts have been measured. Parameterization of these rates has enabled the development of a nucleofugality scale (NFB), shown to quantify and predict the leaving group ability of a range of other Lewis bases. Additivity observed across a number of series R′3-nRnX (X = P, N; R′ = aryl, alkyl) has allowed the formulation of related substituent parameters (nfPB, nfAB), providing a means of calculating NFB values for a range of Lewis bases that extends far beyond those experimentally derived. The utility of the nucleofugality parameter is explored by the correlation of the substituent parameter nfPB with the hydrolyses rates of a series of alkyl and aryl MIDA boronates under neutral conditions. This has allowed the identification of MIDA boronates with heteroatoms proximal to the reacting center, showing unusual kinetic lability or stability to hydrolysis.
Reduction of phosphine oxides into the corresponding phosphines using PhSiH3 as a reducing agent and Ph3C+[B(C6F5)4]? as an initiator is described. The process is highly efficient, reducing a broad range of secondary and tertiary alkyl and arylphosphines, bearing various functional groups in generally good yields. The reaction is believed to proceed through the generation of a silyl cation, which reaction with the phosphine oxide provides a phosphonium salt, further reduced by the silane to afford the desired phosphine along with siloxanes. (Figure presented.).
The metal-free reduction of a range of phosphine(V) oxides employing oxalyl chloride as an activating agent and hexachlorodisilane as reducing reagent has been achieved under mild reaction conditions. The method was successfully applied to the reduction of industrial waste byproduct triphenylphosphine(V) oxide, closing the phosphorus cycle to cleanly regenerate triphenylphosphine(III). Mechanistic studies and quantum chemical calculations support the attack of the dissociated chloride anion of intermediated phosphonium salt at the silicon of the disilane as the rate-limiting step for deprotection. The exquisite purity of the resultant phosphine(III) ligands after the simple removal of volatiles under reduced pressure circumvents laborious purification prior to metalation and has permitted the facile formation of important transition metal catalysts.
The present invention relates to a process for the conversion of a tertiary phosphine oxide to the corresponding tertiary phosphine. The process has desired characteristics such as inexpensiveness, ease of handling, high efficiency, high yield and high conversion, as well as low environmental impact.
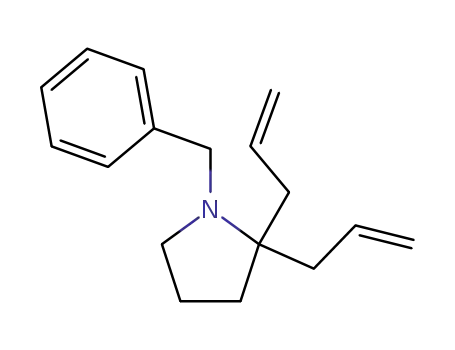
1-benzyl-2,2-di(2-propenyl)pyrrolidine

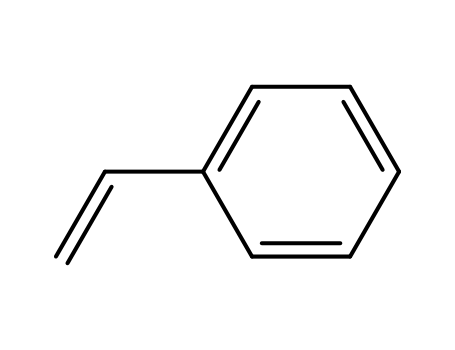
styrene


dibenzyl ether
![1-benzyl-10-azaspiro[4.4]non-7-ene](/upload/2023/2/c5112024-7357-4db8-8ffd-5ff14370197e.png)
1-benzyl-10-azaspiro[4.4]non-7-ene
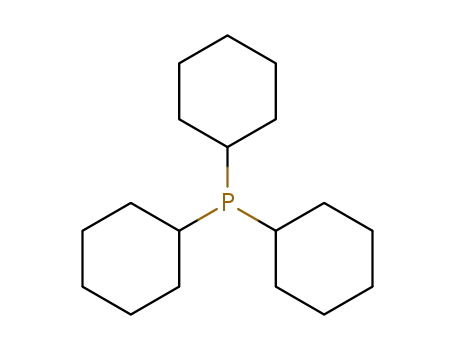
tricyclohexylphosphine
| Conditions | Yield |
|---|---|
|
In
chloroform;
for 7h;
Heating;
|
13% 24% 10% 4% |
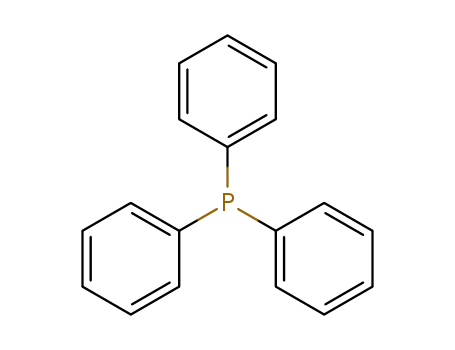
triphenylphosphine
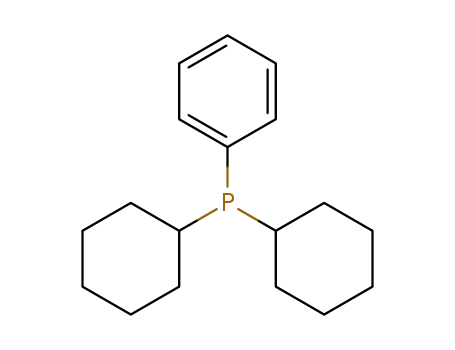
dicyclohexylphenylphosphine
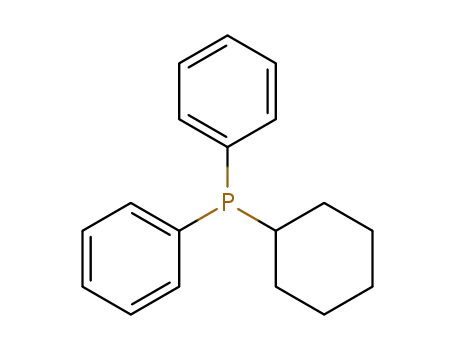
cyclohexyldiphenylphosphine
tricyclohexylphosphine
| Conditions | Yield |
|---|---|
|
With
n-butyllithium; hydrogen;
bis(2,6-diisopropylphenoxy) trichloroniobium(V);
In
hexane; benzene;
at 60 ℃;
under 62057.8 Torr;
Kinetics;
var. of catalyst;
|
|
|
With
n-butyllithium; hydrogen;
|
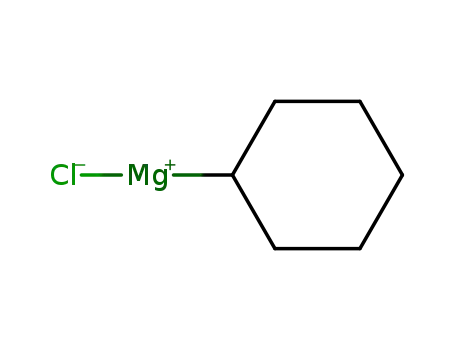
cyclohexylmagnesiumchloride
cyclohexylmagnesium bromide
tricyclohexyl(dithiocarboxylato)phosphonium
triphenylphosphine

butyl-tricyclohexyl-phosphonium; iodide

tricyclohexylmethylphosphonium iodide
tricyclohexylphosphine oxide
tricyclohexylphosphine sulfide

CAS:6299-16-7
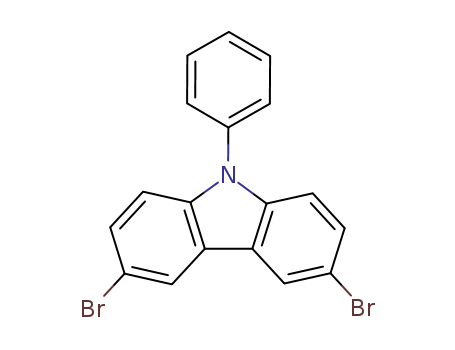
CAS:57103-20-5
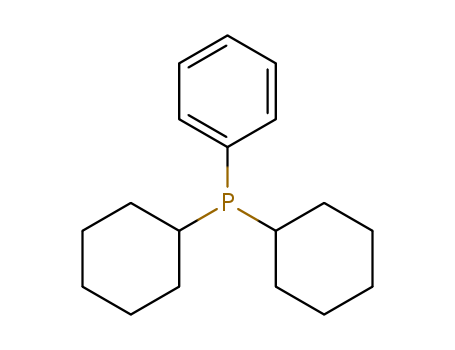
CAS:6476-37-5

CAS:58656-04-5
Molecular Formula:C18H34BF4P
Molecular Weight:368.2
