Your Location:Home >Products >13029-09-9
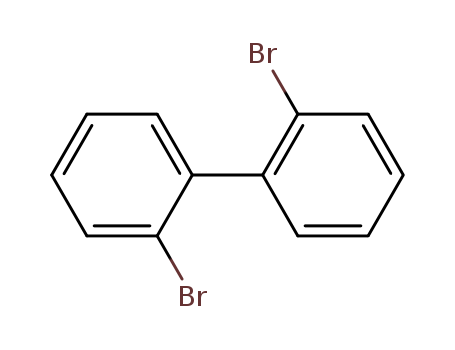
Product Details
Chemical Properties
white crystalline powder
Uses
2,2'-Dibromobiphenyl is used to produce 5,5-dimethyl-5H-dibenzosilole at the temperature of -78 - 20°C. It will need reagent n-BuLi and solvents diethyl ether, hexane.
The utilization of light and inexpensive catalysts to afford hydrogen represents a huge challenge. Following our interest in silicon-containing [FeFe]-hydrogenase ([FeFe]-H2ase) mimics, we report a new model approach for a photocatalytic [FeFe]-H2ase mimic 1, which contains a 1-silafluorene unit as a photosensitizer. Thereby, the photoactive ligand is linked to the [2Fe2S] cluster through S-CH2-Si bridges. Photochemical H2 evolution experiments were performed and revealed a turnover number (TON) of 29. This is the highest reported photocatalytic efficiency for an [FeFe]-H2ase model complex in which the photosensitizer is covalently linked to the catalytic center. We report a viable synthetic pathway for the construction of a new photoactive model of the [FeFe]-hydrogenase ([FeFe]-H2ase) active site with a dithiolate bridge and 1-silafluorene as a photosensitizer. The [FeFe]-H2ase mimic represents a very compact, easily accessible, and inexpensive photocatalyst for hydrogen generation. Copyright
Two novel heterofluorene motifs consisting of the group 14 elements Si and Ge were developed as robust molecular platforms for the construction of organic light-emitting diode (OLED) materials. These two compounds exhibited similar photophysical properties, thermal stabilities and electrochemical behaviors except for the charge transport abilities. The red OLED hosted by a 9-silafluorene derivative (DPS) with Ir(MDQ)2(acac) as the emitter exhibited state-of-the-art current efficiency, power efficiency, and external quantum efficiency (EQE) with maxima of 50.7 cd A-1, 44.7 lm W-1, and 28.3%, respectively. In addition, the maximum EQE of a 9-germafluorene derivative (DPG)-based red device could reach 20%. These results indicated the great potential of group 14 element derivatives in the construction of highly efficient OLED materials.
This paper presents the results of our investigations on the arylation of thiophene using the transition-metal-free “aryne coupling” methodology. The reaction was studied by both experiment and computation (density functional theory) and comparison with phenyllithium was established. In parallel, the effects of the ligand and the salt on the coupling reaction were examined. The results underline the remarkable effect of such additives on the coupling reaction and the potency of the method to construct hetaryl–aryl backbones which open up promising access to a wide range of heterobiaryl structures using the novel “Het-Aryne” route.
The reaction of 2,2'-dibromo-biphenyl on a Ag(111) surface leads to the formation of planar acenes with a high-regioselectivity rather than nonplanar saddle-shaped tetraphenylene as the typical product in solution chemistry. The regioselective aryl-aryl coupling reaction is attributed to the hydrogen repulsion between the reactants on the confined two-dimensional surface.
Lithiation of biphenyl with 2.4 mol of n-butyllithium in the presence of TMEDA led directly to the 2,2'-dilithio derivative (I) in modest, but preparatively useful yields.I, in turn, was converted to a variety of products.The activation of the 2'-position of 2-lithiobiphenyl was shown directly by a separate experiment.MNDO calculations indicate stabilization in I by double bridging and in 2-lithiobiphenyl by intramolecular ? interaction of Li with the o-phenyl group.Similar interactions in substitution transition states rationalize the specificity of the reactions observed.
-
The mechanism of aggregation-induced emission, which overcomes the common aggregation-caused quenching problem in organic optoelectronics, is revealed by monitoring the real time structural evolution and dynamics of electronic excited state with frequency and polarization resolved ultrafast UV/IR spectroscopy and theoretical calculations. The formation of Woodward–Hoffmann cyclic intermediates upon ultraviolet excitation is observed in dilute solutions of tetraphenylethylene and its derivatives but not in their respective solid. The ultrafast cyclization provides an efficient nonradiative relaxation pathway through crossing a conical intersection. Without such a reaction mechanism, the electronic excitation is preserved in the molecular solids and the molecule fluoresces efficiently, aided by the very slow intermolecular charge and energy transfers due to the well separated molecular packing arrangement. The mechanisms can be general for tuning the properties of chromophores in different phases for various important applications.
An efficient method for the homocoupling of aryl halides by electron-transfer oxidation of Lipshutz cuprates (Ar2Cu(CN)Li 2) with organic electron acceptors is disclosed. Thus, various types of Lipshutz cuprates are prepared by successive treatment of aryl or heteroaryl bromides with tert-butyllithium and CuCN. The electron-transfer oxidation of Lipshutz cuprates with p-benzoquinones proceeds smoothly to afford the corresponding homocoupling products in moderate to good yields. Furthermore, it can be applied to the construction of either thiophene- or benzene-fused 10-membered ring cyclophanes. For the synthesis of 10-membered cyclophanes, the linear C-Cu-C structure of Lipshutz cuprates should be maintained in the dimetallacyclic intermediates, producing the large ring cyclophanes efficiently. The X-ray analysis of the cyclophanes reveals that the difference in the bridging atoms results in the different conformations of the macrocyclic rings. Thus, the silicon-bridged cyclophane 5a adopts a D2-symmetric structure with a twisted rhombic arrangement of four thiophene rings, whereas the methylene- and oxygen-bridged cyclophanes 5b and 5c possess C2h- and C2-symmetric structures with chair- and boatlike conformations, respectively. The 1H NMR spectrum of C2-symmetric 5c is temperature-dependent, and the activation energy (ΔG?) for the conformational change is 10.1 kcal/mol.
The Pd-catalyzed homodimerization with respect to arylsulfonyl chlorides as an efficient method for the synthesis of biaryls has been developed. This desulfonylative reaction which was performed at reflux in 1,4-dioxane for 4 h under air afforded the desired products in good to excellent yields. The Pd(OAc)2/PPh3-catalyzed desulfonylative homodimerization of arenesulfonyl chlorides as an efficient method for the synthesis of biaryls was described. The corresponding products were obtained in good to excellent yields.
Thermolysis of 9-azido-9-borafluorene in heptane solution produces the tetramer of a BN-phenanthryne. The isolation of the self-trapping product provides evidence for the involvement of the BN-aryne in the thermolysis reaction. Its formation may be rationalized by denitrogenation of the azide and ring enlargement. Making a first appearance: A BN-aryne analogous to 9,10-phenanthryne can be formed by thermolysis of the 9-azido-9-borafluorene and can undergo cyclotetramerization.
While many foldamer systems reliably fold into well-defined secondary structures, higher order structure remains a challenge. A simple strategy for the organization of folded subunits in space is to link them together within a macrocycle. Previous work has shown that o-phenylenes can be co-assembled with rod-shaped linkers into twisted macrocycles, showing an interesting synergy between folding and thermodynamically controlled macrocyclization. In these systems the foldamer units were largely decoupled from each other both conformationally and electronically. Here, we show that hydrocarbon macrocycles, with very short ethenylene linkers, can be assembled from o-phenylenes using olefin metathesis. Characterization by NMR spectroscopy, X-ray crystallography, and ab initio calculations shows that the products are approximately triangular trimer macrocycles with helical o-phenylene corners in a heterochiral configuration. Their photophysics are dominated by the 4,4'-diphenylstilbene moieties, the longest conjugated segments, with further conjugation broken by the twisting of the o-phenylenes.
Herein we report cyclobuta[1,2-b:3,4-b′]diphenazine (CBDP), a new π-electron molecular scaffold containing two phenazine moieties connected by a four-membered ring. With properly positioned silylethynyl substituting groups, CBDP offers a chromophore with
To learn from Nature how to create an efficient hydrogen-producing catalyst, much attention has been paid to the investigation of structural and functional biomimics of the active site of [FeFe]-hydrogenase. To understand their catalytic activities, the μ
Typically, Suzuki couplings used in polymerizations are performed at raised temperatures in inert atmospheres. As a result, the synthesis of aromatic materials that utilize this chemistry often demands expensive and specialized equipment on an industrial scale. Herein, we describe a bimetallic methodology that exploits the distinct reactivities of palladium and copper to perform high yielding aryl-aryl dimerizations and polymerizations that can be performed on a benchtop under ambient conditions. These couplings are facile and can be performed by simple mixing in the open vessel. To demonstrate the utility of this method in the context of polymer synthesis: polyfluorene, polycarbazole, polysilafluorene, and poly(6,12-dihydro-dithienoindacenodithiophene) were created at ambient temperature and open to air.
An efficient palladium-catalyzed tandem reaction for the one-pot synthesis of 9 H -carbazoles under microwave irradiation is developed. This approach involves a sequential Buchwald-Hartwig amination and a direct arylation from affordable and inexpensive anilines and 1,2-dihaloarenes. For the development of this purpose, a novel and magnetically recoverable palladium nanocatalyst supported on a green biochar under ligand-free conditions is used. Compared to other existing palladium-based protocols, the present synthetic methodology shows a drastic reduction in reaction times and excellent compatibility with different functional groups allowing to obtain a small library of 9 H -carbazoles in high yields and with good regioselectivity. This procedure represents the first example in the direct synthesis of carbazoles using a heterogeneous palladium nanocatalyst from commercial precursors. To examine the application of this protocol, a direct and scalable synthesis of the bioactive carbazole alkaloid clausenalene from commercially available starting materials is described.
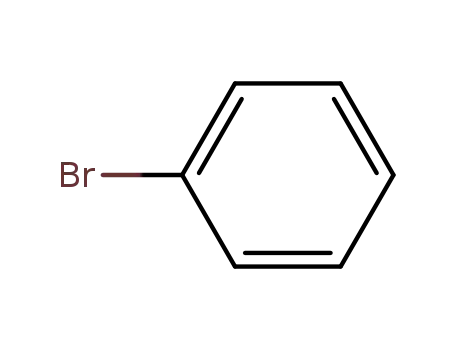
bromobenzene

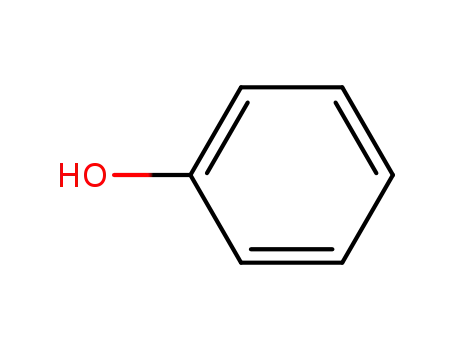
phenol

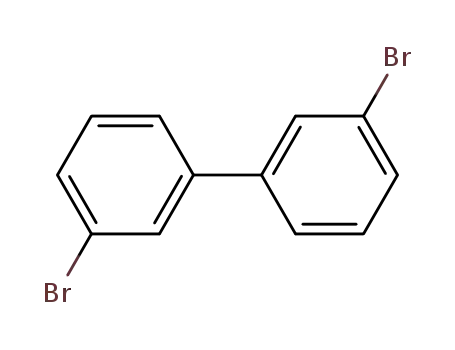
3,3'-dibromobiphenyl
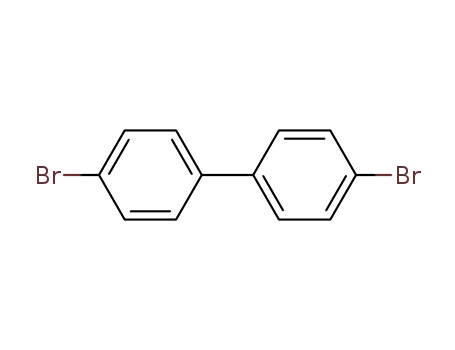
4-(4-bromophenyl)bromobenzene
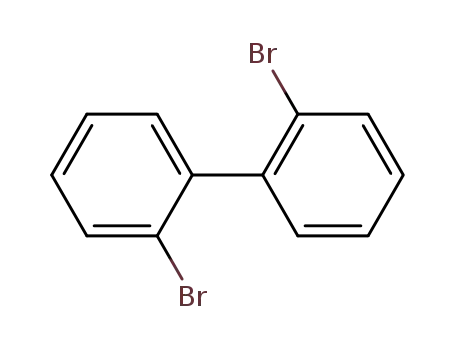
2,2'-dibromobiphenyl
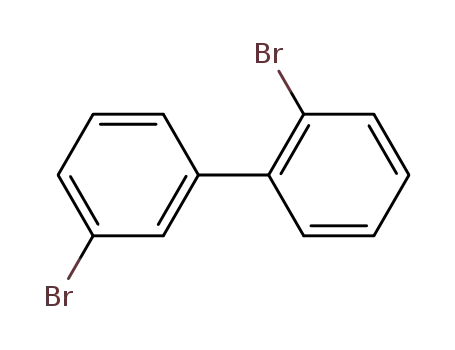
2,3′-dibromo-1,1′-biphenyl
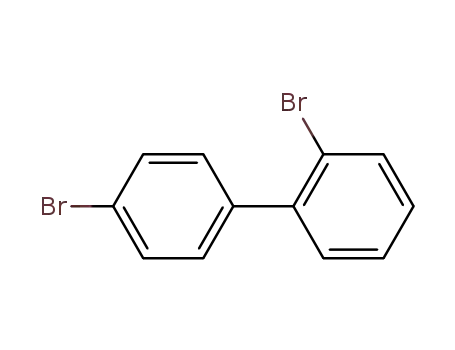
2-bromo-4'-bromobiphenyl
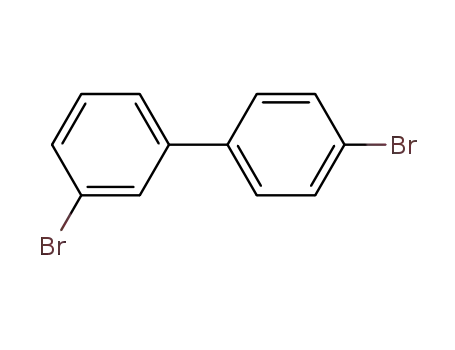
3,4’-dibromobiphenyl
| Conditions | Yield |
|---|---|
|
With
tert.-butylhydroperoxide; iodine;
at 289.9 ℃;
for 0.0272222h;
Product distribution;
|
12.5% 30.9% 16% 17% 19.3% 4.3% |
bromobenzene
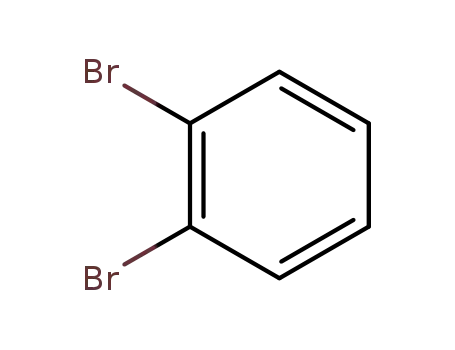
2,3-dibromobenzene
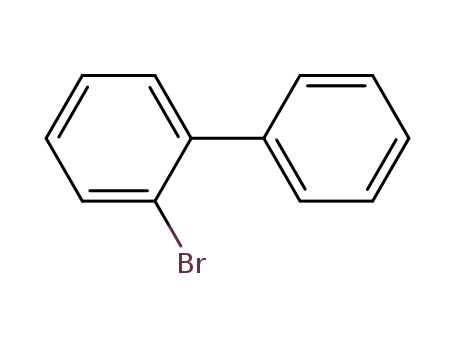
2-Bromobiphenyl
2,2'-dibromobiphenyl
| Conditions | Yield |
|---|---|
|
bromobenzene;
With
tert.-butyl lithium;
In
tetrahydrofuran;
at -80 ℃;
for 1h;
Inert atmosphere;
2,3-dibromobenzene;
In
tetrahydrofuran;
at -50 ℃;
for 2h;
Inert atmosphere;
|
44% 9% |
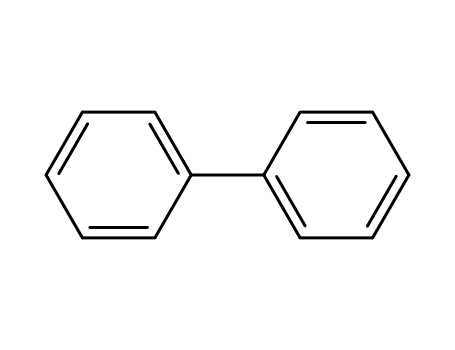
biphenyl
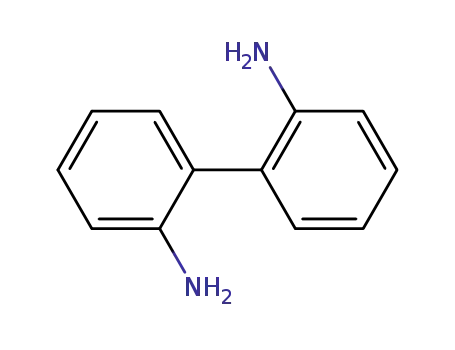
1,1'-biphenyl-2,2'-diamine
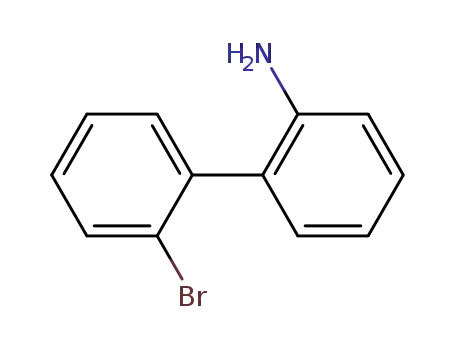
2’-bromo-[1,1’-biphenyl]-2-amine
2,3-dibromobenzene
biphenyl
2-Bromobiphenyl
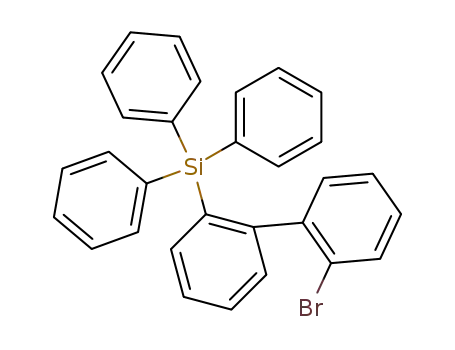
(2'-bromo-biphenyl-2-yl)-triphenyl-silane
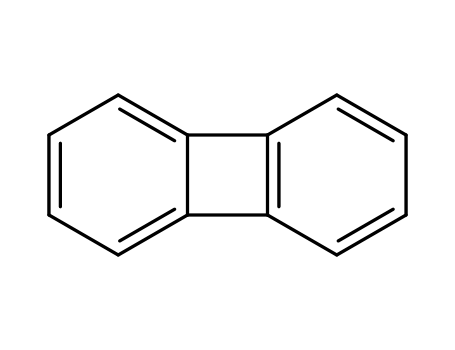
bisbenzocyclobutene
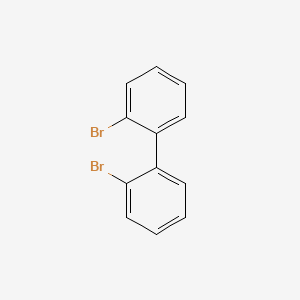
CAS:13029-09-9
Molecular Formula:C<sub>12</sub>H<sub>8</sub>Br<sub>2</sub>
Molecular Weight:312

CAS:125143-53-5
CAS:25550-51-0
